Причины
Возникновение фокальной корковой дисплазии происходит еще при внутриутробном развитии плода. Патология обусловлена нарушением окончательного формирования церебральной коры. В результате этого в головном мозге появляются участки с атипичными структурами. Мозговые ткани отличаются утолщенностью, извилины становятся более плоскими.
Патологический процесс развивается примерно за месяц-полтора до рождения ребенка.
Спровоцировать неправильно развитие тканей мозга могут факторы, которые влияют на будущую маму во время вынашивания ребенка, к примеру, вредные привычки, радиация, гормональные сбои и прочее.
Виды патологии
Фокальная корковая дисплазия бывает трех типов:
- Первый. Наблюдается очаговое изменение архитектоники коры головного мозга.
- Второй. Выявляется локальное нарушение цитоархитектоники, при котором появляются атипичные нейроны и баллонные клетки. Такой вид патологии поражает в большинстве случаев левую или правую лобную долю.
- Третий. Вторичная форма изменений архитектоники коры мозга, возникающая как следствие других болезней, к примеру, склероза, энцефалита, инфекционных патологий.
Какой конкретно тип развивается у пациента, доктор определяет при обследовании.
Симптомы
Фокальная корковая дисплазия проявляется в основном приступами эпилепсии. Они характеризуются кратковременностью, так как длятся меньше минуты. Иногда встречаются ложные приступы.
Также у пациентов наблюдается спутанность сознания после окончаний эпилепсии. Возможны внезапные падения, нарушение координации движений.
Диагностика
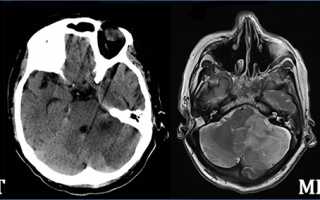
Для выявления фокальной корковой дисплазии применяют магнитно-резонансную томографию. МРТ осуществляется по специальной методике. Она помогает обнаружить даже минимальные изменения в структуре коры головного мозга. Для правильной расшифровки результатов обследования требуется помощь опытного и квалифицированного рентгенолога.
Также больным назначают электроэнцефалографию. Она позволяет выявить эпилептическую активность головного мозга, как в момент приступа, так и вне него.
Для определения участка, где начинается приступ эпилепсии, используют с помощью ПЭТ, которое совмещают с томографией. При проведении такого обследования больному вводят специальный препарат сразу же после первого пароксизмального разряда.
 Читайте также:
Читайте также:

Лечение
Для лечения фокальной корковой дисплазии используют противосудорожные препараты. Они помогают предотвращать приступы эпилепсии. Но бывает так, что данные медикаменты отказываются недостаточно сильными и не справляются со своей задачей. В этом случае принимается решение о проведении оперативного вмешательства.
В зависимости от того, какая область головного мозга поражена, и насколько распространен патологический процесс, применяют одну из следующих методов хирургического вмешательства:
- Селективная резекция эпилептогенной области.
- Классическая резекция височной доли или иных частей головного мозга.
- Тэйлорированная резекция.
После операции нередко наблюдаются стойкие нарушения неврологического характера. Точных мер по предотвращению развития фокальной кортикальной дисплазии тейлора нет, потому что неизвестно, что именно вызывает это заболевание. Чтобы при внутриутробном развитии не возникало никаких нарушений, будущим мамам следует внимательно следить за своим здоровьем.
Дисплазия головного мозга – что это такое
Ограниченное церебральное повреждение на ограниченном участке провоцирует разнообразные клинические проявления. Эпилептические приступы сочетаются с нарушением сознания, кортикальными нарушениями.
Первоначальные признаки нозологии с помощью электроэнцефалографии. Небольшие церебральные изменения устраняются антиконвульсивными препаратами.
Разновидности фокальной дисплазии:
- Первый тип – изменение корковой архитектоники локальное;
- Второй тип – очаговые цитоархитектонические изменения;
- Третий тип – патология архитектоники при вторичных болезнях (темпоральный склероз, церебральная мальформация, энцефалит Расмуссена, ишемия).
Способы лучевой нейровизуализации помогают верифицировать стадию нозологии.
Поликистозная дисплазия мозга характеризуется наличием множества кистозных разрастаний внутри церебральной ткани.

-
Читайте также:

Для диагностики пороков сравнивают результаты КТ и МРТ
Аномалии развития ствола головного мозга – причины возникновения
В зависимости от морфологических изменений выделяют группы аномалий мозга:
- Положения;
- Количества;
- Размера и формы;
- Структуры (строения).
Первая группа нозологий возникает по причине недоразвитие зачатка церебральной структуры или полного отсутствия эмбрионального зачатка. Если ребенок рождается нормальным, прогноз является неблагоприятным из-за отсутствия части мозга.
К аномалиям положения следует отнести удвоение органа, слияние между собой нескольких частей одновременно.
Дефекты положения церебральных структур
Все нозологические формы группы определяются тремя факторами:
- Инверсионное смещение органа относительно собственной оси, срединного положения;
- Дистопия – необычная локализация эмбриональных структур;
- Гетеротопия – патология закладки органа.
Степень смещения определяется выраженность клинических симптомов, длительность жизни человека.
Пороки размеров и формы мозга
Перечень нозологий данной категории определяется рядом морфологических изменений:
- Синостозы нескольких органов;
- Гиперплазия — увеличение количества тканей и размеров церебральных тканей;
- Гипоплазия диспластическая – уменьшение размеров структуры;
- Гипоплазия простая – нет изменений морфологии.
Дефекты строения головного мозга
Нозология сопровождается аномалиями естественного образования отверстия, морфологических особенностей структуры. Гетероплазия развивается на этапе внутриутробного развития. Характеризуется атипичным формированием ткани.

Дисплазия – патология соотношения суставных поверхностей.
Гамартрия – неправильное развитие тканевых структур. Стенотическое сужение канала, протока – бывает врожденным и приобретенным.
Дизонтогенетическая киста сопровождается значительным сужением компенсаторных возможностей органа.
Классификация пороков эмбрионального развития:
- Фетопатии;
- Эмбриопатии;
- Бластопатии;
- Гаметопатии.
В зависимости от времени появления дефектов эмбрионального развития возникает определенный тип нарушений.
По обширности повреждения выделяют виды церебральных дефектов:
- Множественные – поражают сразу несколько мозговых областей;
- Системные – локализуются в пределах одного участка;
- Изолированные – обеспечивают повреждение одного органа.
Врожденные аномалии центральной нервной системы могут провоцироваться инфекционными агентами:
- Токсоплазма;
- Цитомегаловирус;
- Вирус Коксаки.
Встречаются алкогольные аномалии, если беременная женщина употребляла спирт во время вынашивания плода. Патология провоцируется хромосомными аберрациями, генетическими мутациями во время формирования нервной трубки (третья-четвертая неделя беременности).
Основные виды мозговых дефектов
Выделяют дефекты формы, размеров, расположения отдельных анатомических структур. Рассмотрим основные виды врожденных аномалий развития головного мозга.
-
Читайте также:

Что такое энцефалоцеле
Проникновение церебральных тканей через дефекты черепа обуславливает разные неврологические симптомы, зависящие от особенностей участка выпадения. Небольшая анэнцефалия напоминает кефалогематому, но рентгенография черепа выявляет незаращение по средней линии, ассиметричные участки.
С помощью хирургического вмешательства удается устранить патологию, но крупные очаги нельзя устранить эктопированием выпячивания. Энцефалоцеле верифицируется методами лучевой нейровизуализации – МРТ и КТ.
Особенности анэнцефалии
Патология характеризуется отсутствием отдельных костей черепа. Места разрушения зарастают соединительной тканью, что не позволяет оптимально регулировать внутричерепное давление.
Большая часть нозологических форм не совместима с жизнью. Смертельный исход возникает сразу после рождения, когда раскрываются легкие, начинается подача кислорода к церебральной паренхиме.
Проявления микроцефалии
Недоразвитие церебральных тканей формируется у одного ребенка на пять тысяч новорожденных детей. Определяется нозология по уменьшению размера черепной коробки, нарушением соотношения между мозговой и лицевой частью черепа.
Микроцефалия (синдром Джакомини) развивается внутриутробно у женщин с инфекциями, паразитарными болезнями.
Причины первичной микроцефалии:
- Генетические аномалии с передачей по аутосомно-рецессивному типу;
- Токсоплазмоз, энцефалит цитомегаловирус, краснуха.
Этиологические факторы вторичной микроцефалии:
- Церебральные кисты;
- Обызвествления и кровоизлияния внутрь мозговой паренхимы.
Уменьшением размеров черепа характеризуется около десяти процентов олигофрений. С ранних лет у ребенка прослеживается отставание развития, закрытые пороки развития. Умственная отсталость сопровождается судорожным синдромом. Мышечные подергивания сопровождаются неправильным развитием мозговой части черепной коробки.
Чем характеризуется макроцефалия
Диагностировать патологию позволяет увеличение черепной коробки. Нозология характеризуется гипертрофией одного полушария, непропорциональным развитием одной половины. Умственное недоразвитие – самое частое проявление. Судорожные припадки прослеживаются примерно у девяти-десяти процентов пациентов.
Клиника нозологии появляется в течение первых лет жизни, что позволяет своевременно верифицировать патологию. Мозговые миграционные нарушения слишком серьезны для эффективного лечения болезни.
Симптоматика голопрозэнцефалии
Голопрозэнцефалия – болезнь сопровождается дефектом развития полушарий. Отсутствие разделения между церебральными половинами обуславливает изменения активности функциональных центров.
Сильные дисплазии приводят к аномалиям желудочков, асимметричностью лицевой и мозговой части. Выраженные дефекты приводят к омертвению церебральной паренхимы с высокой вероятностью гибели в первые сутки после появления ребенка на свет.
Образование единой полусферы является врожденным пороком из-за генетической аномалии тринадцатой-пятнадцатой хромосом. Нозология нередко сочетается с другими пороками развития:
- Циклопия;
- Этмоцефалия;
- Цебоцефалия.
Заболевание сопровождается мертворождением. Способность к выживанию минимальна. Прогноз неблагоприятный.
Клиника дисплазии церебральной кистозной
Множественные кистозные полости обуславливают миграционные нарушения. Дефекты развития сопровождаются аномалиями распределения спинномозговой жидкости. Многочисленные кисты нельзя удалить. Нередко провоцируют мышечные судороги. Низкая эффективность антиконвульсивного лечения приводит к прогрессированию симптомов.
Единичные кисты могут не представлять опасности. Возможно субклиническое протекание симптоматики на фоне увеличения внутричерепной гипертензии.
Чем проявляется агирия
Лиссэнцефалия – это дефекты формирования архитектоники мозговой коры. Тяжелые судороги обуславливает недоразвитие церебральных извилин. Нозология формирует моторные и психические проявления. Неврологические признаки заболевания – синдром Леннокса-Гасто, Веста.
Слаженность головного мозга провоцирует параличи, парезы, полиморфные судороги. Признаки нозологии развиваются на первом году жизни. Примерно данный промежуток времени живет малыш.
Клинические признаки пахигирии
Дефект развития обусловлен отсутствием формирования вторичных и третичных извилин. Выпрямление борозд второго типа нарушает церебральную архитектонику.
Патология послойного строения коры характеризуется гетеротопией нервных клеток. Хорошо показывает пахигирию МРТ.
Клинические симптомы краниостеноза
Болезнь характеризуется сужением черепа с компрессией головного мозга между костями. В зависимости от прогрессирования выделяют декомпенсированный и компенсированный вариант нозологии.
По особенностям течения выделяют стабильную и прогрессирующую формы болезни. Чаще причины обусловлены ранним зарастанием коронарного или сагиттального швов. Патология без своевременного лечения приводит к летальному исходу, так как появляется ущемление головного мозга. Клинические симптомы зависят от преимущественной локализации зоны сдавления белого вещества и паренхимы.
Неврологическая симптоматика характеризуется многочисленными расстройствами на фоне увеличения внутричерепного давления.
У ребенка с краниостенозом сильная головная боль, поэтому малыш беспокоен, раздражителен, плаксив. Потеря памяти, нарушение концентрации внимания возникает при длительном сохранении состояния. Прогноз патологии неблагоприятный.
Показатели агенезии мозолистого тела
Нозология характеризуется недоразвитием мозолистого тела. Сопутствующая патология – недоразвитие третьего желудочка мозга. Гипоплазия провоцирует недоразвитие мышечной мускулатуры, парезы и параличи, мышечные судороги.
Проявления синдрома Айкарди возникают при сочетании недоразвития мозолистого тела с хориоретинальными пороками. Клиническая картина дополняется миоклоническими судорогами, формированием многочисленных лакунарных узлов в сетчатке глаза, диске зрительного нерва. Нозология характеризуется микрофтальмом, маятникообразными движениями глаз.
Некоторые исследователи выявляют у пациентов с агенезией мозолистого тела дефекты Х-хромосомы у мужчин.
Клиника микрополигирии
Заболевание возникает по причине формирование многочисленных мелких извилин. Нормальная церебральная кора имеет шесть слоев. При аномалии прослеживается четыре слоя. Аномальная структура приводит к клиническим симптомам:
- Расстройство глотания;
- Патология жевательных, мимических мышц;
- Олигофрения;
- Параплегия лица.
Дебют болезни наблюдается на первом году жизни.
Клинические проявления гетеротопии
Нозология возникает из-за нейрональной миграции. Отсутствие передачи нервного сигнала возникает из-за гетеротопионов – патологические скопления в виде ленточной или узловой формы.
Из-за гетеротопии появляется олигофрения, эпилептический синдром, разные мышечные судороги.
Диагностика врожденных пороков головного мозга
Большинство нозологических форм обнаруживается вначале по клиническим проявлениям. Легкое течение, гипотонические сокращения мускулатуры провоцируют судорожный синдром у детей первого года жизни.
Исключить гипоксические и травматологические проявления позволяют инструментальные способы диагностики – УЗИ, нейросонография, МРТ и КТ. Процедур достаточно для выявления аномалий развития, кист, гетеротопических участков.
Электроэнцефалография обнаруживает зоны повышенной церебральной активности при присутствии судорожных подергиваний мышц. Врожденные виды требуют генетической диагностики для исследования ДНК, определения мутаций хромосомного аппарата.
Дисплазия головного мозга – что это такое
Ограниченное церебральное повреждение на ограниченном участке провоцирует разнообразные клинические проявления. Эпилептические приступы сочетаются с нарушением сознания, кортикальными нарушениями.
Первоначальные признаки нозологии с помощью электроэнцефалографии. Небольшие церебральные изменения устраняются антиконвульсивными препаратами.
Разновидности фокальной дисплазии:
- Первый тип – изменение корковой архитектоники локальное;
- Второй тип – очаговые цитоархитектонические изменения;
- Третий тип – патология архитектоники при вторичных болезнях (темпоральный склероз, церебральная мальформация, энцефалит Расмуссена, ишемия).
Способы лучевой нейровизуализации помогают верифицировать стадию нозологии.
Поликистозная дисплазия мозга характеризуется наличием множества кистозных разрастаний внутри церебральной ткани.
Для диагностики пороков сравнивают результаты КТ и МРТ
Аномалии развития ствола головного мозга – причины возникновения
Каждая оболочка головного мозга выполняет определенную функцию. Прозрачная перегородка является мозговым веществом и состоит из двух пластин.
Агенезия – одна из составляющих большинства врожденных пороков головного мозга. Отсутствие перегородки обусловлено неправильным формированием или недоразвитием мозолистого тела.
Агенезия полости прозрачной перегородки относят к аномалиям центральной нервной системы и встречается довольно редко. Характеризуется данная патология полным или частичным отсутствием полости прозрачной перегородки. Данная аномалия достаточно не изучена. Развивается порок уже на второй недели после зачатия.
Предрасполагающие факторы к развитию агенезии:
- Наследственность
- Мутации
- Внутриутробные инфекции
- Недостаточное поступление питательных веществ к плоду
Также на развитие патологии влияют токсические вещества и лекарственные препараты, которые женщина принимала в период беременности. Способствовать врожденным аномалиям головного мозга может употребление таких препаратов, как Триметадион, Фенитоин, Изотретиноин и некоторые другие. Эти лекарственные средства, применяемые в первом триместре беременности, оказывают влияние на формирование головного мозга и могут привести к дефектам развития.
Если будущая мама употребляла алкоголь, то у ребенка развивается фатальный алкогольный синдром. Это также предрасполагает к врожденной патологии. Инфекции матери или полученные травмы на 12-22 недели беременности могут привести к нарушениям в развитии мозга плода.
Агенезия прозрачной перегородки не возникает изолированно. Обычно патология является частью различных церебральных аномалий: агенезия мозолистого тела, порэнцефалия, гидранэнцефалия, септо-оптическая дисплазия, голопрозэнцефалия и др.
Симптомы в тяжелой форме выявляются в детском возрасте в течение первых двух лет жизни. При рождении дети с агенезией выглядят здоровыми и развиваются нормально до трех месяцев. На данном этапе развития появляются первые признаки патологии.
При агенезии наблюдаются следующие симптомы:
- Появление порэнцефалии
- Микроэнцефалия
- Недостаточное формирование извилины
- Синдром Айкарди
- Атрофия зрительных и слуховых нервов

Также на фоне патологии может наблюдаться раннее половое созревание, приступы и припадки. Проявляться патология может по-разному. При частичной агенезии указанные признаки могут не наблюдаться у ребенка и не влиять на развитие, однако нужно будет регулярно наблюдаться у невролога. Иногда агенезия может протекать без клинических проявлений на протяжении нескольких лет.
В зависимости от морфологических изменений выделяют группы аномалий мозга:
- Положения;
- Количества;
- Размера и формы;
- Структуры (строения).
Первая группа нозологий возникает по причине недоразвитие зачатка церебральной структуры или полного отсутствия эмбрионального зачатка. Если ребенок рождается нормальным, прогноз является неблагоприятным из-за отсутствия части мозга.
К аномалиям положения следует отнести удвоение органа, слияние между собой нескольких частей одновременно.
Все нозологические формы группы определяются тремя факторами:
- Инверсионное смещение органа относительно собственной оси, срединного положения;
- Дистопия – необычная локализация эмбриональных структур;
- Гетеротопия – патология закладки органа.
Степень смещения определяется выраженность клинических симптомов, длительность жизни человека.
Перечень нозологий данной категории определяется рядом морфологических изменений:
- Синостозы нескольких органов;
- Гиперплазия — увеличение количества тканей и размеров церебральных тканей;
- Гипоплазия диспластическая – уменьшение размеров структуры;
- Гипоплазия простая – нет изменений морфологии.
Дефекты строения головного мозга
Нозология сопровождается аномалиями естественного образования отверстия, морфологических особенностей структуры. Гетероплазия развивается на этапе внутриутробного развития. Характеризуется атипичным формированием ткани.
Дисплазия – патология соотношения суставных поверхностей.
Гамартрия – неправильное развитие тканевых структур. Стенотическое сужение канала, протока – бывает врожденным и приобретенным.
Дизонтогенетическая киста сопровождается значительным сужением компенсаторных возможностей органа.
Классификация пороков эмбрионального развития:
- Фетопатии;
- Эмбриопатии;
- Бластопатии;
- Гаметопатии.
В зависимости от времени появления дефектов эмбрионального развития возникает определенный тип нарушений.
По обширности повреждения выделяют виды церебральных дефектов:
- Множественные – поражают сразу несколько мозговых областей;
- Системные – локализуются в пределах одного участка;
- Изолированные – обеспечивают повреждение одного органа.
Врожденные аномалии центральной нервной системы могут провоцироваться инфекционными агентами:
- Токсоплазма;
- Цитомегаловирус;
- Вирус Коксаки.
Встречаются алкогольные аномалии, если беременная женщина употребляла спирт во время вынашивания плода. Патология провоцируется хромосомными аберрациями, генетическими мутациями во время формирования нервной трубки (третья-четвертая неделя беременности).
Диагностика врожденных пороков головного мозга
Большинство нозологических форм обнаруживается вначале по клиническим проявлениям. Легкое течение, гипотонические сокращения мускулатуры провоцируют судорожный синдром у детей первого года жизни.
Исключить гипоксические и травматологические проявления позволяют инструментальные способы диагностики – УЗИ, нейросонография, МРТ и КТ. Процедур достаточно для выявления аномалий развития, кист, гетеротопических участков.
Электроэнцефалография обнаруживает зоны повышенной церебральной активности при присутствии судорожных подергиваний мышц. Врожденные виды требуют генетической диагностики для исследования ДНК, определения мутаций хромосомного аппарата.
Диагностируется патология во 2-3 триместре беременности. В пренатальный период выявить аномалии головного мозга довольно сложно, так как плод может принять такое положение, что не позволит четко рассмотреть все структуры мозга. Определить аномалию можно на УЗИ с 18 недели беременности, не раньше. После рождения для подтверждения диагноза назначают энцефалографию, УЗИ, КТ, МРТ.
Исследовать структуры полости черепа у детей с рождения при возможной агенезии можно с помощью нейросонографии. Проводится процедура через открытый родничок. Благодаря нейросонографии можно исследовать структуры мозга. С помощью УЗИ можно обнаружить патологию в головном мозге даже в том случае, если она не сопровождается симптомами.
Данный метод дополняет нейросонографию и позволяет выявить отсутствие перегородки, изменение структуры желудочков головного мозга. Магнитно-резонансная томография помогает определить характер поражения, отсутствие прозрачной перегородки и другие патологии, которые невозможно определить на УЗИ. Если агенезия частичная, то выявить патологию еще труднее.
Лечение и прогноз
Не существует определенных методик лечения для коррекции агенезии мозолистого тела и на его фоне отсутствие прозрачной перегородки. В основном лечение заключается в устранении серьезных симптомов, проявляющиеся при агенезии у ребенка, или в уменьшении их проявления. Из медикаментозных препаратов назначают антиэпилептические средства, бензодиазепины, кортикостероидные гормоны. Однако даже консервативное лечение может не принести положительных результатов.
Из кортикостероидных гормонов используют преимущественно Преднизолон и Дексаметазон. Пациентам с врожденной патологией назначают Фенобарбитал. Это средство относится к противосудорожным. Бензодиазепины являются психоактивными веществами, которые воздействую на центральную нервную систему, и оказывают седативное, снотворное, анксиолитическое действие.
Полезное видео — УЗИ головного мозга у новорожденного.
Два передние желудочки мозга разделяются прозрачной перегородкой. С её помощью мозолистое тело прикрепляется к своду черепа. При отсутствии перегородки столб лежит на дне переднего желудочка. По размеру он больше, чем вместе взятых 2 передних желудочка.
У некоторых пациентов с агенезией перегородки не наблюдалась предрасположенность к умственным расстройствам и неврологическим нарушениям.
Если аномалия развивается самостоятельно и не сопровождается другими патологиями, то прогноз в большинстве случаев благоприятный. Дети с такой патологией развиваются практически нормально или имеют некоторые проблемы в неврологическом развитии. Однако в случае развития различных аномалий головного мозга прогноз неблагоприятный.
Основные виды мозговых дефектов
Выделяют дефекты формы, размеров, расположения отдельных анатомических структур. Рассмотрим основные виды врожденных аномалий развития головного мозга.
Проникновение церебральных тканей через дефекты черепа обуславливает разные неврологические симптомы, зависящие от особенностей участка выпадения. Небольшая анэнцефалия напоминает кефалогематому, но рентгенография черепа выявляет незаращение по средней линии, ассиметричные участки.
С помощью хирургического вмешательства удается устранить патологию, но крупные очаги нельзя устранить эктопированием выпячивания. Энцефалоцеле верифицируется методами лучевой нейровизуализации – МРТ и КТ.
Патология характеризуется отсутствием отдельных костей черепа. Места разрушения зарастают соединительной тканью, что не позволяет оптимально регулировать внутричерепное давление.
Большая часть нозологических форм не совместима с жизнью. Смертельный исход возникает сразу после рождения, когда раскрываются легкие, начинается подача кислорода к церебральной паренхиме.
Недоразвитие церебральных тканей формируется у одного ребенка на пять тысяч новорожденных детей. Определяется нозология по уменьшению размера черепной коробки, нарушением соотношения между мозговой и лицевой частью черепа.
Микроцефалия (синдром Джакомини) развивается внутриутробно у женщин с инфекциями, паразитарными болезнями.
Причины первичной микроцефалии:
- Генетические аномалии с передачей по аутосомно-рецессивному типу;
- Токсоплазмоз, энцефалит цитомегаловирус, краснуха.
Этиологические факторы вторичной микроцефалии:
- Церебральные кисты;
- Обызвествления и кровоизлияния внутрь мозговой паренхимы.
Уменьшением размеров черепа характеризуется около десяти процентов олигофрений. С ранних лет у ребенка прослеживается отставание развития, закрытые пороки развития. Умственная отсталость сопровождается судорожным синдромом. Мышечные подергивания сопровождаются неправильным развитием мозговой части черепной коробки.
Диагностировать патологию позволяет увеличение черепной коробки. Нозология характеризуется гипертрофией одного полушария, непропорциональным развитием одной половины. Умственное недоразвитие – самое частое проявление. Судорожные припадки прослеживаются примерно у девяти-десяти процентов пациентов.
Клиника нозологии появляется в течение первых лет жизни, что позволяет своевременно верифицировать патологию. Мозговые миграционные нарушения слишком серьезны для эффективного лечения болезни.
Голопрозэнцефалия – болезнь сопровождается дефектом развития полушарий. Отсутствие разделения между церебральными половинами обуславливает изменения активности функциональных центров.
Сильные дисплазии приводят к аномалиям желудочков, асимметричностью лицевой и мозговой части. Выраженные дефекты приводят к омертвению церебральной паренхимы с высокой вероятностью гибели в первые сутки после появления ребенка на свет.
Образование единой полусферы является врожденным пороком из-за генетической аномалии тринадцатой-пятнадцатой хромосом. Нозология нередко сочетается с другими пороками развития:
- Циклопия;
- Этмоцефалия;
- Цебоцефалия.
Заболевание сопровождается мертворождением. Способность к выживанию минимальна. Прогноз неблагоприятный.
Множественные кистозные полости обуславливают миграционные нарушения. Дефекты развития сопровождаются аномалиями распределения спинномозговой жидкости. Многочисленные кисты нельзя удалить. Нередко провоцируют мышечные судороги. Низкая эффективность антиконвульсивного лечения приводит к прогрессированию симптомов.
Единичные кисты могут не представлять опасности. Возможно субклиническое протекание симптоматики на фоне увеличения внутричерепной гипертензии.
Лиссэнцефалия – это дефекты формирования архитектоники мозговой коры. Тяжелые судороги обуславливает недоразвитие церебральных извилин. Нозология формирует моторные и психические проявления. Неврологические признаки заболевания – синдром Леннокса-Гасто, Веста.
Слаженность головного мозга провоцирует параличи, парезы, полиморфные судороги. Признаки нозологии развиваются на первом году жизни. Примерно данный промежуток времени живет малыш.
Дефект развития обусловлен отсутствием формирования вторичных и третичных извилин. Выпрямление борозд второго типа нарушает церебральную архитектонику.
Патология послойного строения коры характеризуется гетеротопией нервных клеток. Хорошо показывает пахигирию МРТ.
Болезнь характеризуется сужением черепа с компрессией головного мозга между костями. В зависимости от прогрессирования выделяют декомпенсированный и компенсированный вариант нозологии.
По особенностям течения выделяют стабильную и прогрессирующую формы болезни. Чаще причины обусловлены ранним зарастанием коронарного или сагиттального швов. Патология без своевременного лечения приводит к летальному исходу, так как появляется ущемление головного мозга. Клинические симптомы зависят от преимущественной локализации зоны сдавления белого вещества и паренхимы.
Неврологическая симптоматика характеризуется многочисленными расстройствами на фоне увеличения внутричерепного давления.
У ребенка с краниостенозом сильная головная боль, поэтому малыш беспокоен, раздражителен, плаксив. Потеря памяти, нарушение концентрации внимания возникает при длительном сохранении состояния. Прогноз патологии неблагоприятный.
Нозология характеризуется недоразвитием мозолистого тела. Сопутствующая патология – недоразвитие третьего желудочка мозга. Гипоплазия провоцирует недоразвитие мышечной мускулатуры, парезы и параличи, мышечные судороги.
Проявления синдрома Айкарди возникают при сочетании недоразвития мозолистого тела с хориоретинальными пороками. Клиническая картина дополняется миоклоническими судорогами, формированием многочисленных лакунарных узлов в сетчатке глаза, диске зрительного нерва. Нозология характеризуется микрофтальмом, маятникообразными движениями глаз.
Некоторые исследователи выявляют у пациентов с агенезией мозолистого тела дефекты Х-хромосомы у мужчин.
Заболевание возникает по причине формирование многочисленных мелких извилин. Нормальная церебральная кора имеет шесть слоев. При аномалии прослеживается четыре слоя. Аномальная структура приводит к клиническим симптомам:
- Расстройство глотания;
- Патология жевательных, мимических мышц;
- Олигофрения;
- Параплегия лица.
Дебют болезни наблюдается на первом году жизни.
Нозология возникает из-за нейрональной миграции. Отсутствие передачи нервного сигнала возникает из-за гетеротопионов – патологические скопления в виде ленточной или узловой формы.
Из-за гетеротопии появляется олигофрения, эпилептический синдром, разные мышечные судороги.
Причины возникновения заболевания
Возникновение ФКД обуславливается внутриутробными нарушениями в развитии церебральной коры, что обуславливает нарушения миграции клеток мозговой оболочки, образуя участок с аномально развитыми нейронами. Формируется ФКД на конечной стадии беременности, приблизительно за шесть недель до родов.
Из-за того, что у некоторых групп пациентов при обследовании были найдены рецессивные дефекты в структуре гена TSC1, нельзя полностью исключить генетическую причину развития заболевания. Большинство современных научных исследований ведется именно в этом направлении.
Усилия ученых сконцентрированы на разработке методик:
- Раннего (внутриутробного) выявления дефектного гена – данная разработка позволит выявить предрасположенность ребенка к возникновению заболевания в будущем.
- Выявления возможности передачи дефектного гена ребенку от одного из родителей – данная разработка позволит выявить риск передачи гена потомству еще на этапе планирования беременности.
- Замены дефектного гена – данная разработка позволит эффективно лечить заболевание (даже на ранних стадиях), что позволит избежать использования травматичного и чреватого развитием тяжелейших осложнений оперативного лечения.
Виды
До недавнего времени медицинская наука выделяла всего два базовых вида дисплазии, однако, в начале 2011 года был принят новый классификатор, предполагающий деление ФКД на три базовых типа.
Согласно данному классификатору ФКД бывает:
- Первого типа. Характеризуется возникновением одного или нескольких локальных дефектов архитектоники коры головного мозга.
- Второго типа. Характеризуется развитием очаговых поражений архитектоники, с наличием аномалий в развитии нейронов и клеточных балок. В большинстве случаев поражает лобную долю.
- Третьего типа. Развитие дефектов архитектоники спровоцировано негативным воздействием какой–либо мозговой патологии.
Симптомы заболевания
Главным симптомом, опираясь на который можно диагностировать наличие у пациента корковой дисплазии является – краткосрочные эпилептические приступы. Манифестирует заболевания рано, часто еще до достижения пациентом 5 лет.
Приступы заболевания сложные (сопровождающиеся расстройством поведенческих реакций), интенсивные (осложняются потерей равновесия и возникновением спонтанных двигательных реакций) и кратковременные (редко длятся дольше одной минуты). В раннем возрасте при отсутствии должного лечения провоцируют развитие аутизма.
Симптоматика ФКД напрямую зависит от ее типа, локализации, выраженности симптомов, наличии осложнений и заболеваний, провоцирующих ее появление. Ранняя манифестация в большинстве случаев отягчается задержкой психического и умственного развития.
Симптомы дисплазии первого типа менее выражены, однако, в некоторых случаях данный тип ФКД может провоцировать ухудшение когнитивных способностей у пациента.
Симптомы дисплазии второго типа намного более выражены, чем первого. ФКД данного типа сопровождается возникновением тяжелейших эпилептических припадков.
Симптомы дисплазии третьего типа напрямую зависят от характера основного заболевания.
Диагностика заболевания
Главным диагностическим методом, позволяющим определить наличие ФКД, является – МРТ диагностика, которая, однако, должна производиться согласно специальному диагностическому протоколу. Толщина среза, согласно которому не должна превышать полутора миллиметров. Только столь тщательное сканирование позволяет выявить даже самые минимальные изменения в коре.
В процессе диагностики значение имеет квалификация и опыт врача рентгенолога, из-за чего интерпретация результатов должна вменяться в обязанность исключительно специалисту.
Проведение МРТ исследования позволяет выявить следующие признаки наличия ФКД:
- Изменение толщины коры, в месте предполагаемой локализации дисплазии.
- Отсутствие четко выраженного перехода между белым и серым мозговым веществом.
- Изменение хода мозговых извилин.
Каждый вид ФКД имеет спектр характерных именно для него симптомов. Уточнить данные полученные при прохождении МРТ позволяет проведение электроэнцефалографии, позволяющей выявить наличие локальной эпилептической активности, не только в момент эпилептического припадка, но и в период после него. Во время приступа отмечается активация участков мозга, расположенных в зонах локализации дисплазии. Данная закономерность объясняется наличием за пределами основной локализации дисплазии аномальных мозговых клеток.
Также обозначить зоны локализации дисплазии можно при помощи ПЭТ диагностики, дополненной МРТ исследованием. Однако у данного диагностического метода существует один значительный нюанс, ПЭТ исследование позволяет локализовать дисплазию только в период эпилептического припадка (контрастное вещество должно быть введено в организм пациента исключительно после первого пароксизмального заряда). Данный вид диагностики особенно ценен, в случае если МРТ диагностика не дала четких и однозначных результатов. Для получения более точного результата может применяться электрокортикография.
Терапия
Лечение больного курируется врачом неврологом, совместно с врачом эпилептологом. Лечение начинается с выбора и определения эффективной дозировки противосудорожного препарата. С данными целями может использоваться карбамазепин, диазепам, леветирацетам, топирамат и препарат вальпроевой кислоты. В большинстве случаев эпилепсия, отягощенная ФКД, оказывается крайне устойчивой к какому-либо противосудорожному лечению.
В указанном выше случае должен быть поставлен вопрос о хирургической резекции пораженного участка. Оперативное лечение проводится исключительно врачом нейрохирургом, совместно с неврологом. В связи с тем, что дисплазия носит фокальный характер, оперативное лечение является эффективным.
Многие специалисты настаивают на целесообразности как можно более полного и радикального удаления пораженного мозгового участка. Данное утверждение спорно, потому что локализация клеток, расположенных вокруг основного очага поражения достаточно обширна, а их полное удаление без нанесения значительного вреда пациенту невозможно.
В зависимости от тяжести симптомов, их локализации и размера пораженного участка может быть использован один из трех вариантов хирургического лечения:
- Селективное удаление зоны локализации дисплазии.
- Стандартное удаление пораженной части головного мозга.
- Точечное удаление зоны поражения корковой дисплазией.
При ФКД третьего типа целесообразно удаление, как самого участка дисплазии, так и главного очага поражения.
Прогноз развития заболевания
Прогноз развития заболевания напрямую зависит от типа дисплазии, качества проведенной терапии, а также успешности радикального лечения. В большинстве случаев консервативное лечение дисплазии не приносит эффекта.
Возникновение заболевания в детском возрасте может спровоцировать проявление отклонений в развитии, с постепенным развитием олигофрении.
Оперативное лечение является результативным, при одиночном очаге поражения. Практически у половины пациентов возобновления заболевания не наблюдается. Однако через десять лет после оперативного вмешательства стойкая ремиссия наблюдается только у трети пациентов. Скорее всего, рецидив заболевания в данном случае связывается с неполным удалением поврежденного сегмента.
Возникновение рецидива заболевания непосредственно после проведенного оперативного вмешательства наблюдается приблизительно у двух пациентов их ста, а в случае распространенного поражения, рецидив наступает у шести пациентов из ста прооперированных.
Риск рецидива увеличивается, в случае если оперативное вмешательство проводилось вблизи функционально важных участков головного мозга.