Синдром Лея (также называемый болезнью Ли, подострой некротизирующей энцефаломиелопатией) – наследственное нейрометаболическое расстройство, которое поражает центральную нервную систему.

Назван в честь Арчибальда Дени Ли, британского психоневролога, который впервые описал состояние в 1951 году. Обнаруживаются нормальные уровни тиамина- монофосфата- дифосфата, но уровень тиамина трифосфата снижается или отсутствует. Считается, что расстройство вызвается блокировкой фермента тиаминдифосфаткина.
Признаки и симптомы
Симптомы синдрома Лея начинаются в младенчестве и приводят к смерти в течение нескольких лет. Признаки могут появиться в любом возрасте, в том числе в подростковом или взрослом. Пациенты могут жить много лет после постановки диагноза.
Симптомы впервые появляются после запускающего события, которое обременяет выработку энергии организмом, такого как инфекция или операция. Общее течение синдрома Лея – эпизодическая регрессия развития во время метаболического стресса. У некоторых пациентов длительные периоды без прогрессирования заболевания, у других развивается прогрессирующее снижение.

У детей с синдромом есть такие симптомы, как диарея, рвота, дисфагия (проблемы с глотанием или сосанием), что приводит к неспособности развиваться. Дети с ранней болезнью раздражительны, плачут гораздо чаще, чем обычно. Часто наблюдаются судороги. Избыток лактата обнаруживается в моче, спинномозговой жидкости, крови.
По мере прогрессирования заболевания мышечная система истощается по всему телу, так как мозг не может контролировать сокращение мышц. Гипотония (низкий мышечный тонус, сила), дистония (непроизвольное, устойчивое сокращение мышц), атаксия(отсутствие контроля над движением) часто наблюдаются.
Особенно чувствительны мышцы, которые контролируют глаза, становятся слабыми, парализованными, неконтролируемыми в условиях, называемых офтальмопарезом (слабость, паралич), нистагмом (непроизвольные движения глаз).
Медленные саккады иногда видны. Сердце и легкие также поражаются в результате болезни Leigh. Гипертрофическая кардиомиопатия (утолщение части сердечной мышцы) иногда встречается и может привести к смерти. Асимметричная гипертрофия перегородки связана с синдромом Лея. Характерны – высокий лоб, большие уши, аномалии лица не встречаются.
 Читайте также:
Читайте также:

Дыхательная недостаточность является наиболее распространенной причиной смерти. Другие неврологические симптомы включают периферическую невропатию, потерю чувствительности в конечностях, вызванную повреждением периферической нервной системы.
Гипертрихоз наблюдается при синдроме Лея, вызванном мутациями в ядерном гене SURF1.
Мнение врача:
Синдром Лея, также известный как синдром внезапной детской смерти, представляет собой состояние, при котором младенец внезапно умирает во сне, обычно в возрасте до одного года. Врачи считают, что этот синдром может быть связан с проблемами дыхания или нервной системы, но конкретные причины до сих пор не установлены. Синдром Лея является серьезной угрозой для детей, и важно соблюдать меры предосторожности, такие как спать на спине, избегать перегревания и использовать безопасные спальные места для младенцев. Родители должны обратиться к врачу для получения дополнительной информации и рекомендаций по предотвращению этого опасного состояния.
Геномика
Мутации в митохондриальной ДНК (мтДНК), более 30 генов ядерной ДНК (ген SURF1) вовлечены в болезнь Лея.
Нарушения окислительного фосфорилирования, процесс, посредством которого клетки производят основной источник энергии аденозинтрифосфата (АТФ), от мутаций либо в мтДНК, либо генах, закодированных в ядре.
Последние составляют большую часть болезни, хотя не всегда возможно определить конкретную мутацию, ответственную за состояние у конкретного человека. Четыре из пяти белковых комплексов участвующие в окислительном фосфорилировании чаще всего нарушаются, либо из-за неправильно сформированного белка, либо из-за ошибки сборки этих комплексов.
Независимо от генетической основы, приводит к неспособности затронутых мутацией, выполнять свою роль в окислительном фосфорилировании. Поражаются важные клетки ствола мозга, базальных ганглиев. Это вызывает хронический недостаток энергии в клетках, их гибели, влияет на центральную нервную систему, подавляет двигательные функции.
Сердце, другие мышцы требуют много энергии, подвержены клеточной гибели, вызванной хроническим дефицитом энергии.
Митохондриальные мутации ДНК
Функция митохондрий заключается в преобразовании потенциальной энергии глюкозы, аминокислот, жирных кислот в аденозинтрифосфат (АТФ) при процессе, называемом окислительным фосфорилированием.
Митохондрии несут собственную ДНК, называемую митохондриальной (мтДНК). Информация, там хранящаяся, используется для производства нескольких ферментов, необходимых для производства АТФ.
От 20 до 25 процентов случаев синдрома Лея – мутации митохондриальной ДНК.
Передается по материнской линии детям мужского и женского пола, но отцы не могут передавать митохондриальные гены.
-
Читайте также:

Опыт других людей
Синдром Лея – это серьезное заболевание, вызванное вирусом, который может привести к осложнениям у детей. Родители выражают беспокойство по поводу возможных последствий этого заболевания. Они отмечают, что важно обратить внимание на профилактику и своевременное лечение, чтобы защитить своих детей от опасных последствий.
Мутации ядерной ДНК
Ядерная ДНК включает большую часть генома организма. Наследуется от обоих родителей. Синдром Лея, вызванный мутациями ядерной ДНК, наследуется по аутосомно-рецессивному типу. Это означает, что две копии мутантного гена необходимы, чтобы вызвать заболевание.
Два незатронутых родителя, каждый из которых несет по одному мутантному аллелю, могут иметь больного ребенка, если тот наследует мутантный аллель от обоих родителей.
От 75 до 80 процентов синдрома случаются от мутаций ядерной ДНК.
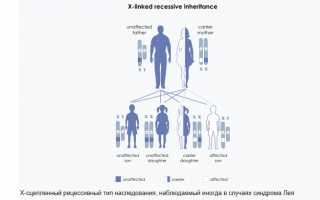
Х-сцепленный синдром Лея
Вызван дефицитом комплекса пируватдегидрогеназы (PDHC), чаще всего с участием субъединицы PDHC, которая кодируется X-связанным геном (OMIM 308930). Неврологические особенности, вызванные дефицитом PDHC, неотличимы от других форм. Однако другие особенности (кроме лактоацидоза) не обнаруживаются при дефиците PDHC.

Х-сцепленный рецессивный синдром Лея затрагивает детей мужского пола гораздо чаще, чем женского, потому что у них есть только одна копия Х-хромосомы. Детям женского пола понадобится две копии дефектного гена, которые будут затронуты.
Французский канадский
Тип, обнаруженный с высокой частотой в регионе Сагене-Лак-Сен-Жан в Квебеке. Вызван мутацией гена LRPPRC, расположенном на маленьком (‘p’) плече хромосомы 2. Этот подтип заболевания впервые описан в 1993 году у 34 детей этого региона. Хотя Субъединицы белка, обнаруженные в пораженных клетках, функциональны, они не правильно собраны.

Дефицит почти полный в тканях мозга, печени и значительный (50% нормальной активности фермента) в фибробластах (клетках соединительной ткани), скелетных мышцах. Ткани почек и сердца не имеют дефицита ЦОГ.
-
Читайте также:

Французский канадский синдром Лея имеет симптомы, аналогичные другим типам. Возраст начала заболевания – 5 месяцев, средний возраст смерти – 1 год и 7 месяцев. Дети с этим заболеванием задерживаются в развитии, имеют слегка дисморфные черты лица, включая гипоплазию средней части и широкий носовой мостик, хронический метаболический ацидоз, гипотонию (снижение мышечной силы).
Другие симптомы:
- тахипноэ (необычно быстрое дыхание);
- плохая способность к сосанию;
- гипогликемия (низкий уровень сахара в крови);
- тремор.
Тяжелый, внезапный метаболический ацидоз является частой причиной смертности.
Генеалогические исследования предполагают, что ответственная мутация привезена в регион ранними европейскими поселенцами.
Патофизиология
Характерные симптомы синдрома Лея частично вызваны двусторонними очаговыми поражениями ствола мозга, базальных ганглий, мозжечка, других областей мозга. Повреждения принимают различные формы, включая зоны демиелинизации, спонгиоза, глиоза, некроза, пролиферации капилляров.
Демиелинизация –потеря миелиновой оболочки вокруг аксонов, подавляя их способность общаться с другими нейронами. Ствол мозга участвует в поддержании основных жизненных функций, таких как дыхание, глотание, кровообращение.
Базальные ганглии и мозжечок контролируют движение и равновесие. Поэтому повреждение приводит к основным симптомам – потере контроля над функциями, контролируемыми этими областями.
Лактоацидоз вызван накоплением пирувата, который не усваивается у людей с определенными типами дефицита окислительного фосфорилирования. Пируват либо превращается в аланин с помощью аланинаминотрансферазы, либо в молочную кислоту с помощью лактатдейдрогеназы. Оба эти вещества накапливаются в организме.
Диагностика
Синдром Лея подтверждается клиническими данными, лабораторными, генетическими исследованиями.
Клинические данные
Дистония, нистагм, проблемы с вегетативной нервной системой предполагают повреждение базальных ганглиев и ствола головного мозга. Другие симптомы также указывают на повреждение головного мозга, такие как гипертрихоз, неврологически вызванная глухота.
-
Читайте также:

Лабораторные результаты лактоацидоза, ацидемии, гипераланинемии (повышенные уровни аланина в крови) указывают на синдром Лея. Оценка уровня органических кислот в моче показывает нарушение метаболического пути.
Дифференциальный диагноз
Другие заболевания имеют сходную клиническую картину. Исключение других причин, сходных клинических симптомов является первым шагом к диагностике.
Состояния, которые похожи:
- перинатальная асфиксия;
- ядерная желтуха;
- отравление угарным газом;
- токсичность метанола;
- дефицит тиамина;
- болезнь Вильсона;
- биотин-чувствительные заболевания базальных ганглиев;
- некоторые формы энцефалита.
Перинатальная асфиксия вызывает двустороннее поражение ганглиев, повреждение таламуса, которые похожи на признаки, наблюдаемые при болезни Ли.
Лечение
Янтарная кислота показала свою эффективность как при синдроме Лея, так и при МЕЛАС. Диета с высоким содержанием жиров и низким содержанием углеводов должна соблюдаться, если вовлечен ген Х-хромосомы.
Тиамин (витамин B) назначают, если известен или подозревается дефицит пируватдегидрогеназы. Симптомы лактоацидоза лечат путем добавления в пищу бикарбоната натрия (пищевой соды) или цитрата натрия, но эти вещества не устраняют причину.
Дихлорацетат эффективен при лечении лактоацидоза, ассоциированного с болезнью Ли; исследования этого вещества продолжаются. Иногда добавки коэнзима Q10 улучшали симптомы.
Клинические испытания препарата EPI-743 для лечения синдрома Лея продолжаются.
Прогноз
Различные генетические причины и типы синдрома Лея имеют разные прогнозы, хотя все они плохие. Самые тяжелые формы заболевания, вызванные полным дефицитом одного из пораженных белков, вызывают смерть в возрасте нескольких лет.
Если дефицит не является полным, прогноз несколько улучшается. Ожидается, что больной ребенок доживет до 6-7 лет, в редких случаях – до подросткового возраста.
Эпидемиология
Встречается у 1 из 40 000 живорождений, хотя в некоторых популяциях этот показатель намного выше. В регионе Сагене-Лак-Сен-Жан центрального Квебека расстройство встречается с частотой 1 на 2000 новорожденных.
Частые вопросы
Что такое Синдром Лея и каковы его основные симптомы?
Синдром Лея – это редкое, но серьезное заболевание, которое характеризуется внезапной потерей сознания у детей в возрасте от 6 месяцев до 5 лет. Основные симптомы включают в себя рвоту, судороги и потерю сознания.
Каковы причины возникновения Синдрома Лея и как его можно предотвратить?
Причины возникновения Синдрома Лея могут быть разнообразны, включая вирусные инфекции, генетические факторы или даже воздействие окружающей среды. Предотвращение этого синдрома включает в себя регулярные медицинские осмотры, вакцинацию от вирусных инфекций и предотвращение травм головы.
Полезные советы
СОВЕТ №1
Обратите внимание на поведение ребенка: если он часто жалуется на боли в области живота, имеет проблемы с пищеварением или наблюдается ухудшение аппетита, стоит обратиться к врачу для диагностики.
СОВЕТ №2
Изучите симптомы Синдрома Лея: рвота, понос, вздутие живота, потеря веса, слабость и задержка в росте у ребенка могут быть признаками этого синдрома.