Болезнь Тея Сакса, амавротическая идиотия является редким, нейродегенеративным расстройством, в котором дефицит фермента (гексозаминидазы А) приводит к чрезмерному накоплению определенных жиров (липидов), известных как ганглиозиды в мозге и нервных клетках.
Это аномальное накопление ганглиозидов приводит к прогрессирующей дисфункции центральной нервной системы.
Оно классифицируется как лизосомальное заболевание. Лизосомы являются основными клетками пищеварения. Ферменты лизосом ломают или «переваривают» питательные вещества, включая некоторые сложные углеводы и жиры.
Описание
Когда фермент, такой как гексозаминидаза А, которые необходимы для разрушения определенных веществ, таких как жиры, отсутствуют или неэффективны, они накапливаются в лизосомале. Это называется ненормальным «хранилищем», когда в лизосоме возникает слишком много жирового материала.

Симптомы, связанные с синдромом Тея Сакса, включают:
- преувеличенную реакцию испуга на внезапные шумы;
- вялость;
- потерю ранее приобретенных навыков (психомоторную регрессию);
- сильно уменьшенный мышечный тонус (гипотония).
По мере прогрессирования болезни у детей развиваются;
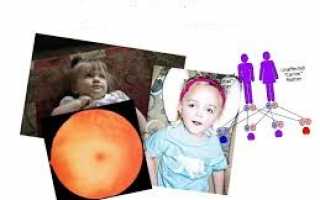
- вишнево-красные пятна в среднем слое глаз;
- происходит постепенная потеря зрения и слуха;
- увеличивается мышечная жесткость;
- прогрессирует ограничение движений (спастичность);
- возможен паралич;
- неконтролируемые электрические нарушения мозга (судороги);
- ухудшение когнитивных процессов (деменция).
Классическая форма болезни Тея-Сакса возникает в младенчестве. Это наиболее распространенная форма, является фатальной в раннем детстве.
Существуют также подростковые и взрослые формы синдрома, но они редки. Дети с подростковой формой, называемой подострой, развивают симптомы позже, чем с инфантильной, и обычно живут дольше.
Взрослая форма, называемая поздним началом болезни, возникает от подросткового возраста до середины 30-ти лет. Симптомы и тяжесть варьируются.
 Читайте также:
Читайте также:

Синдром Тея Сакса наследуется аутосомно-рецессивным способом. Расстройство является результатом изменений (мутаций) гена, известного как
HEXA, который регулирует продукцию фермента гексозаминидазы А. Ген
HEXAотображен на длинной руке (q) хромосомы 15 (15q23-q24). Лечения нет, оно направлено на облегчение конкретных симптомов.
Другое название болезни – это тип ганглиозидоза GM2 типа 1. Существуют два других связанных заболевания, называемых болезнью Сандхоффа и дефицитом активатора гексозаминидазы, которые неотличимы от синдрома Тея-Сакса.
Они могут быть дифференцированы только путем тестирования, определения основной причины. Эти два нарушения также вызывают снижение активности гексозаминидазы, но вызваны изменениями в разных генах. Эти три нарушения известны как ганглиозидозы GM2.
Мнение врача:
Синдром Тея Сакса, амавротическая идиотия, является редким генетическим заболеванием, характеризующимся тяжелым умственным и физическим отставанием. Врачи отмечают, что данное заболевание часто приводит к нарушениям в развитии нервной системы и моторики, что существенно ограничивает возможности пациентов. Однако, благодаря современным методам диагностики и ранней медицинской помощи, удается улучшить качество жизни пациентов с данным синдромом. Врачи подчеркивают важность комплексного подхода к лечению и реабилитации, который позволяет снизить проявление симптомов и обеспечить пациентам максимальную поддержку.
Синонимы
- GM2-ганглиозидоз;
- GM2 ганглиозидоз, тип 1;
- Дефицит альфа-субъединицы гексоамидазы (вариант B);
- Дефицит гексосаминидазы А;
- Недостаток HEXA;
- Сфинголипидоз, Тай-Сакс;
- ТСД.
Стадии
- Инфантильная болезнь Тэя-Сакса;
- Подострое заболевание;
- Поздняя стадия.
Опыт других людей
Синдром Тея Сакса, также известный как амавротическая идиотия, вызывает многочисленные обсуждения среди специалистов и обычных людей. Одни считают его редким и таинственным заболеванием, требующим большего внимания и исследований, другие выражают свою поддержку семьям, сталкивающимся с этим диагнозом. Несмотря на различные точки зрения, важно продолжать освещать эту проблему и обеспечивать поддержку тем, кто сталкивается с этим сложным состоянием.
Признаки
Болезнь Тея-Сакса подразделяется на классическую или инфантильную форму, ювенильную и взрослую или позднего начала. У лиц с инфантильной стадией симптомы появляются в возрасте от трех до пяти месяцев. При поздней форме симптомы видны от подросткового возраста до 30-ти лет.
Infantile форма
Инфантильная форма характеризуется почти полным отсутствием активности фермента гексозаминидазы А. Расстройство прогрессирует быстро, что приводит к значительному психическому и физическому ухудшению.
Младенцы могут казаться совершенно здоровыми при рождении. Начальные симптомы, обычно развиваются между 3 – 6 месяцами, включают:
- слабость мышц;
- подергивание (миоклонические толчки);
- преувеличенную реакцию испуга, например, при возникновении внезапного или неожиданного шума.
Реакция испуга частично вызвана повышенной чувствительностью к звуку (акустическая гиперчувствительность).

-
Читайте также:

В период между 6 – 10 месяцами пораженные дети не приобретают новых моторных навыков. У них нет контакта глазами, наблюдаются необычные движения глаз. Они вялы и раздражительны.
Младенцы медленно растут, имеют прогрессирующую мышечную слабость, слабый мышечный тонус (гипотония), пониженное умственное функционирование.
Могут демонстрировать постепенную потерю зрения, непроизвольные мышечные спазмы, которые приводят к медленным, жестким движениям (спастичности), потерю ранее приобретенных навыков, таких как способность сидеть, ползать.
Характерным симптомом является развитие красных пятен на глазах. Это состояние возникает, когда макулярные клетки ухудшаются, повреждая лежащую в основе сосудистую оболочку.

Возникают поведенческие трудности, прогрессирующая потеря речи, жизненных навыков, интеллектуальных способностей. Иногда появляется вишнево-красное пятно в глазах.
Может возникнуть дегенерация нерва, который несет импульсы от глаза к мозгу, чтобы образовывать образы (атрофия зрительного нерва).
У некоторых встречается пигментный ретинит, большая группа нарушений зрения, вызывающая прогрессирующее дегенерацию сетчатки, светочувствительную мембрану, которая покрывает внутреннюю поверхность глаз. Дети меньше реагируют на окружающую среду.
Поздняя
Симптомы, связанные с поздней стадией амавротической идиотии, варьируются. У затронутых лиц не будет всех признаков, перечисленных ниже. Расстройство прогрессирует гораздо медленнее, чем инфантильная форма.
-
Читайте также:

Изменчивость позднего начала наблюдается в пределах одной семьи. У одного человека могут быть симптомы в возрасте 20 лет, другой получает их в 60- 70-лет как небольшие проблемы с мышцами.
Первоначальные признаки, связанные с поздней стадией синдрома Тея сакса, могут включать:
- неуклюжесть;
- изменения настроения;
- прогрессирующую мышечную слабость;
- амиотрофию.
В зависимости от возраста проявляется тремор, подергивание мышц (фасцикуляции), судороги, невнятная речь (дизартрия), неспособность координировать движения (атаксия), затруднение глотания (дисфагия), дистония.

При инфантильной стадии наблюдается почти полное отсутствие гексозаминидазы A. На поздней стадии заболевания наблюдается дефицит активности. Поскольку существует некоторая активность фермента, расстройство менее выражено и прогрессирует гораздо медленнее.
Точное количество ферментов в позднем начале болезни Тея-Сакса сильно варьируется. Следовательно, возраст начала, тяжести, специфических симптомов и скорости прогрессирования позднего начала также значительно варьируются от одного человека к другому.
Большинство генетических заболеваний определяются по статусу двух копий гена, полученного от отца и одного от матери. Если человек наследует один нормальный ген и один мутированный, то будет носителем заболевания, без проявления симптомов.
- Риск наследования от двух родителей-носителей составляет 25% при каждой беременности.
- Риск иметь ребенка носителя, составляет 50%.
- Шанс на получение ребенком нормальных генов от обоих родителей составляет 25%.
Риск одинаковый для мужчин и женщин.
Затронутые популяции
Болезнь поражает мужчин и женщин в равных количествах. Чаще встречается у еврейского народа ашкеназского происхождения, то есть у восточного или среднеевропейского происхождения. Примерно один из 30 евреев Ашкенази несет измененный ген. Кроме того, один из 300 человек неашкеназского еврейского наследия является носителем.

Сообщалось о заболеваниях людей итальянского, ирландского, французского, канадского происхождения, особенно тех, кто живет в общине Каджун Луизиана и юго-восточном Квебеке. В общей популяции скорость переноса для измененного гена составляет приблизительно 1 из 250-300 человек.
Позднее заболевание происходит реже, чем инфантильная форма. Однако редкие формы расстройства часто остаются непризнанными. Они мало представлены, что затрудняет определение истинной частоты нарушений у населения в целом.
Связанные нарушения
Симптомы следующих расстройств могут быть сходными с симптомами болезни Тея-Сакса. Сравнения полезны для дифференциального диагноза:
Болезнь Сандхофф
Редкое генетическое заболевание, приводящее к прогрессирующему ухудшению центральной нервной системы (нейродегенеративное расстройство). Дефицит ферментов гексозаминидазы А и В приводит к накоплению липидов в головном мозге и других органах тела. Это заболевание классифицируется как лизосомальное.
Наиболее распространенная форма поражает младенцев от трех до шести месяцев. Симптомы включают проблемы с кормлением, общую слабость, преувеличенный рефлекс испуга в ответ на внезапный громкий шум.
Двигательные задержки, потеря ранее приобретенных навыков, умственные нарушения являются прогрессивными. У детей наблюдается потеря слуха, зрения, судороги.
Встречаются очень редкие формы, когда симптомы начинаются позже в детстве, юности или взрослой жизни. Болезнь Сандхоффа является тяжелой формой болезни Тея-Сакса и не ограничивается какой-либо конкретной этнической группой. Вызвана мутациями в гексозаминидазе B (
HEXB), наследуется как аутосомно-рецессивный признак.
Синдром Ли
Редкое генетическое неврометаболическое расстройство. Характеризуется дегенерацией центральной нервной системы (головного, спинного мозга, зрительного нерва). Симптомы начинаются в возрасте от трех месяцев до двух лет, но некоторые пациенты не проявляют признаков несколько лет.
Признаки связаны с прогрессирующим неврологическим ухудшением и включают потерю ранее приобретенных моторных навыков, потерю аппетита, рвоту, раздражительность, судороги. По мере развития синдрома Ли, появляется генерализованная слабость, отсутствие мышечного тонуса (гипотония), эпизоды лактоацидоза, что приводит к нарушению функции органов дыхания и почек.
Несколько различных ферментных дефектов могут вызывать синдром, описанный более 60 лет назад. У большинства людей с синдромом Ли есть дефекты производства митохондрий, такие как дефицит фермента митохондриального дыхательного цепного комплекса или комплекса пируватдегидрогеназы.
У большинства синдром Ли наследуется как аутосомно-рецессивный признак. Однако X-связанное рецессивное и материнское наследование, обусловленное мутацией митохондриальной ДНК, являются дополнительными способами передачи.
Нейронные цероидные липофусцинозы (NCL)
Группа прогрессирующих дегенеративных нейрометаболических заболеваний. Они имеют сходные симптомы и частично отличаются возрастом, при котором появляются. В младенчестве встречаются две формы: классическая инфантильная болезнь CLN1 (болезнь Сантавуори) и болезнь CLN2 (поздний инфантильный тип).
NCL характеризуются аномальным накоплением жирных, гранулированных веществ (например, пигментных липидов, липопигментов, липофусцина) в нервных клетках (нейронах) головного мозга, а также в других тканях организма. Это приводит к прогрессирующему ухудшению (атрофия) определенных участков мозга, неврологическим нарушениям и другим характерным симптомам.
Заболевания лизосомальной области являются наследственными метаболическими. Они характеризуются аномальным накоплением различных токсических материалов в клетках организма из-за дефицита фермента. Влияют на различные части тела, включая скелет, мозг, кожу, сердце, центральную нервную систему.
Симптомы следующих расстройств могут быть сходными с симптомами поздней стадии Тея-Сакса. Сравнения полезны для дифференциального диагноза:
Взрослый нейронный кероидный липофусциноз (ANCL)
Общий термин для нескольких редких генетических нарушений, которые относятся к группе прогрессирующих дегенеративных нейрометаболических заболеваний, известных как нейроновые кероидные липофусцинозы (NCL).
Эти расстройства имеют сходные симптомы и частично отличаются возрастом, при котором появляются. Начало ANCL обычно составляет около 30 лет, но может возникать в подростковом возрасте или у людей старше 50 лет.
НКЛ характеризуются аномальным накоплением жирных, гранулированных веществ (пигментных липидов, цероида, липофусцина) в нейронах и других тканях организма. Характеризуется постепенным ухудшением (атрофией) определенных участков мозга, неврологическими нарушениями.
ANCL иногда называют болезнью Kufs. Исторически болезнь Kufs разбита на тип A или B. ANCLs вызваны изменениями в разных генах и имеют разные признаки и симптомы.
Амиотрофический боковой склероз (ALS)
Характеризуется легкой мышечной слабостью, неуклюжими движениями рук, затруднением выполнения задач, требующих деликатных движений пальцев, рук. Мышечная слабость ног может стать причиной падения.
Люди с БАС испытывают трудности с глотанием (дисфагия), речь замедленна. Дополнительные симптомы включают прогрессирующую слабость губ, потерю функции языка, рта, бульбарные симптомы. Ночные судороги возникают чаще всего в мышцах ног или бедра. Амиотрофический боковой склероз прогрессирует быстро или медленно, постепенно поражая дополнительные мышцы.
У лиц с БАС может наблюдаться неконтролируемое подергивание мышц (фасцикуляция), скованность в ногах, кашель. Поскольку способность двигаться постепенно ухудшается, люди подвергаются повышенному риску дыхательной недостаточности. Точная причина болезни неизвестна.
Диагностика
Диагноз подтверждается тщательной клинической оценкой и специализированными тестами, такими как анализы крови, измеряющие уровни гексозаминидазы А в организме. Хексозаминидаза А снижается у людей с болезнью Тея-Сакса, отсутствует в инфантильной форме.
Молекулярно-генетическое тестирование подтверждает диагноз. Оно обнаруживает мутации гена
HEXA,которые вызывают расстройство.
В некоторых случаях возможно, что диагноз синдрома Тея-Сакса может быть заподозрен до рождения (пренатально) на основе специализированных тестов, таких как амниоцентез, хорионная выборка ворсинок (CVS).
Во время амниоцентеза берется образец жидкости, окружающий развивающийся плод, CVS получает образец ткани из части плаценты. Эти образцы изучают, чтобы определить, присутствует ли гексозаминидаза А так, как у людей с болезнью она отсутствует или ее очень мало. Это называется ферментным анализом.

Анализы крови могут определить, являются ли люди носителями болезни Тея-Сакса (есть одна копия гена). Родственники должны быть проверены, чтобы определить, являются ли они носителями гена болезни. Пары, которые планируют иметь ребенка и имеют еврейское происхождение (не только ашкенази), поощряются к проведению скрининга, перед беременностью.
Лечение
Специального лечения нет. Оно направлено на конкретные симптомы и может потребовать скоординированных усилий группы специалистов. Рекомендуется генетическая консультация пострадавшим людям и их семьям. Психосоциальная поддержка необходима для всей семьи.
Из-за потенциальных трудностей кормления следует следить за состоянием питания и надлежащей гидратацией.
Может потребоваться питательная поддержка. В дополнение к ней иногда необходима питающая трубка, которая поможет предотвратить случайное попадание пищи, жидкости или другого постороннего материала в легкие (аспирация).
Противосудорожные препараты используются для лечения судорог, но не могут быть эффективными для всех. Кроме того, тип и частота приступов часто требует изменения типа лекарства или дозировки.
Исследовательская терапия
Продолжается работа по развитию ферментной заместительной терапии (ERT) для болезни Тей-Сакса. Она заключается в замене фермента у лиц, испытывающих его недостаток или отсутствие.
Синтетические варианты отсутствующих энзимов используются для лечения людей с другими заболеваниями лизосомального хранения, включая синдром Херлера, Фабри, болезнь Гоше. Однако, ERT не оказалась успешной для людей с синдромом тея сакса. Одна из проблем состоит в невозможности преодолеть гематоэнцефалический барьер.
- Генная терапия изучается как возможный подход к терапии некоторых лизосомных нарушений памяти. При генной терапии дефектный ген, заменяется нормальным, позволяя получать активный фермент, который предотвращает развитие и прогрессирование заболевания.
Учитывая постоянную передачу нормального гена, который продуцирует активный фермент во всех очагах заболевания, эта форма терапии, скорее всего, приведет к «излечению». Однако в настоящее время существуют технические трудности для успеха данного подхода.
- Чапероновая терапия также изучается. Этот тип терапии использует очень маленькие молекулы, которые присоединяются к вновь созданным ферментам гексозаминидазы А, прежде чем мутированные разрушаются. Они направляют их к лизосоме, где ферменты выполняют нормальную функцию.
Такая молекула может пересекать гематоэнцефалический барьер. Терапия находится на начальных этапах исследования.
- Препарат под названием пириметамин был применен в качестве лечения болезни Тей-Сакса. Затронутые лица, принимающие этот препарат, показали повышенную активность гексозаминидазы А.
Однако она не привела к заметному улучшению неврологических или психических симптомов. Необходимы дополнительные исследования, чтобы определить, играет ли пириметамин какую-либо роль в лечении этого синдрома.
Частые вопросы
Что такое синдром Тея Сакса, амавротическая идиотия?
Синдром Тея Сакса, амавротическая идиотия – это редкое генетическое заболевание, характеризующееся нарушением развития мозга, нарушением движений и отсутствием речи.
Каковы основные симптомы синдрома Тея Сакса, амавротической идиотии?
Основные симптомы включают в себя нарушения координации движений, умственную отсталость, непроизвольные движения, судороги, а также отсутствие развития речи.
Каковы методы лечения синдрома Тея Сакса, амавротической идиотии?
На сегодняшний день нет специфического метода лечения этого синдрома. Лечение направлено на облегчение симптомов, реабилитацию и поддержание пациента с помощью физической терапии, логопедических занятий и медикаментозной поддержки.
Полезные советы
СОВЕТ №1
Если вы заметили у ребенка или у себя признаки амавротической идиотии, обратитесь к специалисту для проведения диагностики. Чем раньше будет поставлен диагноз, тем быстрее можно начать лечение и поддержку ребенка.
СОВЕТ №2
Ищите информацию у надежных источников, таких как медицинские учреждения, организации, занимающиеся поддержкой людей с амавротической идиотией, и авторитетные медицинские публикации. Это поможет вам получить достоверную информацию о синдроме Тея Сакса и амавротической идиотии.
СОВЕТ №3
Общайтесь с другими родителями и семьями, у которых есть опыт в воспитании детей с амавротической идиотией. Они могут поделиться своими знаниями, опытом и поддержкой, что очень важно в такой ситуации.